Unravelling the Impact of Halogen Bonds in Medicinal Chemistry with QM calculations
Co-Authors: Luboš Remeň, Gabriele Conti, Benedetta Girardi, Cornelia Zumbrunn, Corinna Grisostomi, Martin Bolli, Sven Glutz, Christina Stamm, Daniela Krüsi, Claire Guilois, Aengus Mac Sweeney, Alain Chambovey, Célia Mueller Grandjean, John Gatfield
For the last decade medicinal chemists were challenged by increasingly difficult targets to discover new, potent drugs. These oftentimes deemed “undruggable” targets, nonetheless, share some features that make working on them worthwhile: i) they modulate disease relevant pathways in a way that could lead to new therapies, ii) it can be hard to achieve potent molecules, while keeping the required properties in check and iii) most importantly for a scientist, we are constantly expanding our knowledge to also apply unconventional methods in our day-to-day drug design process.
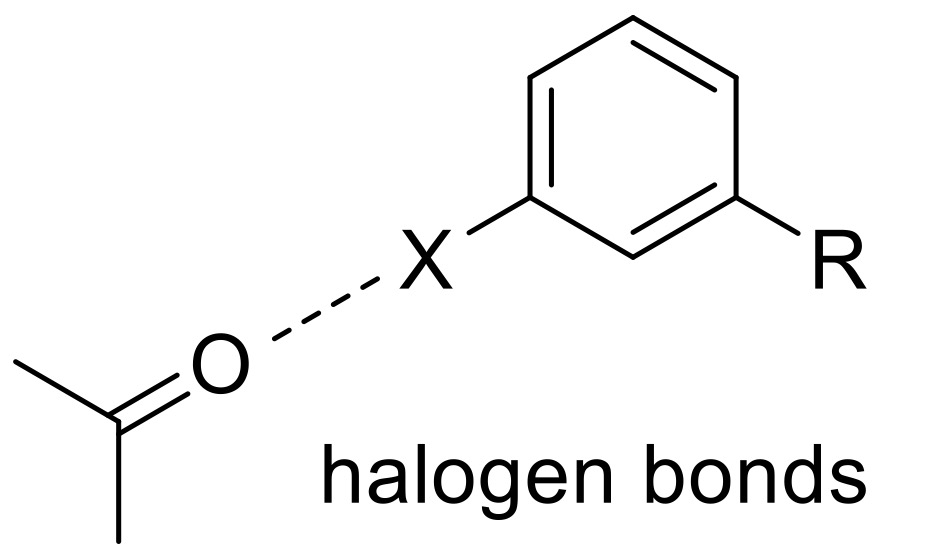
One of these more unconventional methods is to look for uncommon interactions in highly-resolved ligand–protein crystal structures, such as halogen bonds, and use quantum mechanical calculations to uncover different substitutions to increase the halogen bonding potential. Here we describe a specific ligand–protein halogen bond that (significantly) contributes to the ligand–protein interaction, and its incorporation into our ligand design.
[1] C. Bissantz, B. Kuhn, and M. Stahl, J. Med. Chem. 2010, 53, 5061–5084 5061
[2] M. R. Scholfield, C. M. Vander Zanden, M. Carter, and P. Shing Ho, Protein Science 2013 Vol 22:139—152
[3] N. K. Shinada, A. G. de Brevern, P. Schmidtke, J. Med. Chem. 2019, 62, 9341−9356