Microcanonical and finite-temperature ab initio molecular dynamics simulations on quantum computers
Ab initio molecular dynamics (AIMD) is a powerful tool to predict the properties of molecular and condensed matter systems. However, the quality of this procedure relies on the availability of rigorous electronic structure calculations. The development of quantum processors has shown great potential for the efficient evaluation of accurate ground and excited state energies of molecular systems, opening up new avenues for molecular dynamics simulations. In this talk, we address the use of variational quantum algorithms for the calculation of accurate atomic forces to be used in AIMD. In particular, we provide solutions for the alleviation of the statistical noise associated with the measurements of the expectation values of energies and forces, as well as schemes for the mitigation of the hardware noise sources (in particular, gate infidelities, qubit decoherence, and readout errors). Despite the relative large error in the calculation of the potential energy, our results show that the proposed algorithms can provide accurate MD trajectories in the microcanonical (constant energy) ensemble. Furthermore, exploiting the intrinsic noise associated to the quantum measurement process, we also propose a Langevin dynamics algorithm for the simulation of canonical, i.e., constant temperature, dynamics. Both algorithms (microcanonical and canonical) are applied to the simulation of simple molecular systems such as H2 and H3+. Finally, we also provide results for the dynamics of H2 obtained with IBM quantum computer ibmq_athens.
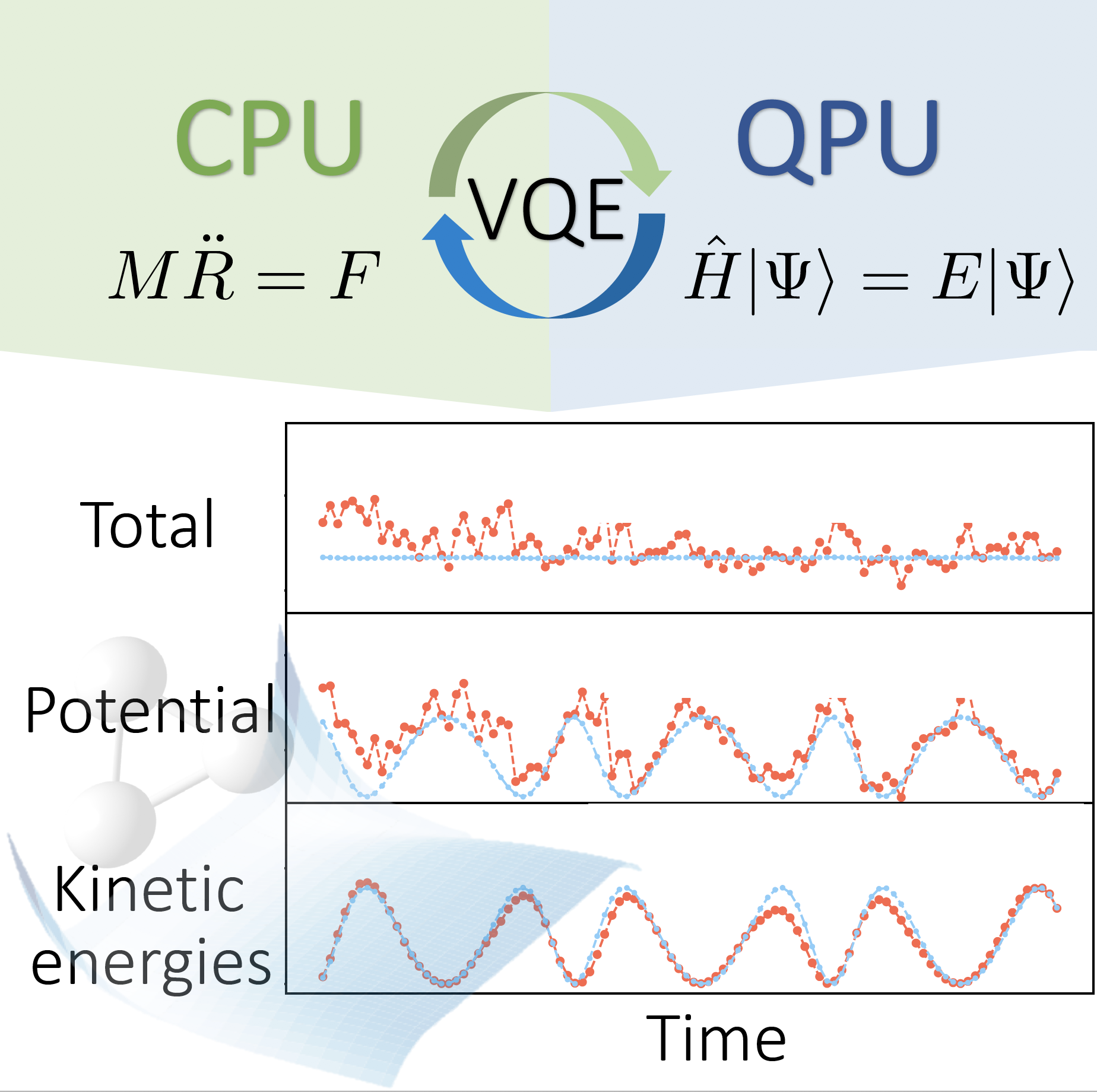
[1] Igor O. Sokolov, Panagiotis Kl. Barkoutsos, Lukas Moeller, Philippe Suchsland, Guglielmo Mazzola, and Ivano Tavernelli, Phys. Rev. Research, 2021, 3, 013125.