A Covalent Strategy to Develop Highly Selective Chemical Probes Targeting PI3Kα
Inhibitors of the phosphatidylinositol 3-kinase (PI3K) – mechanistic target of rapamycin (mTOR) axis are considered as valuable assets in cancer therapy. A considerable effort has been dedicated to the development of drugs targeting the PI3K-mTOR pathway[1-4], and some inhibitors are currently evaluated in preclinical and clinical studies.
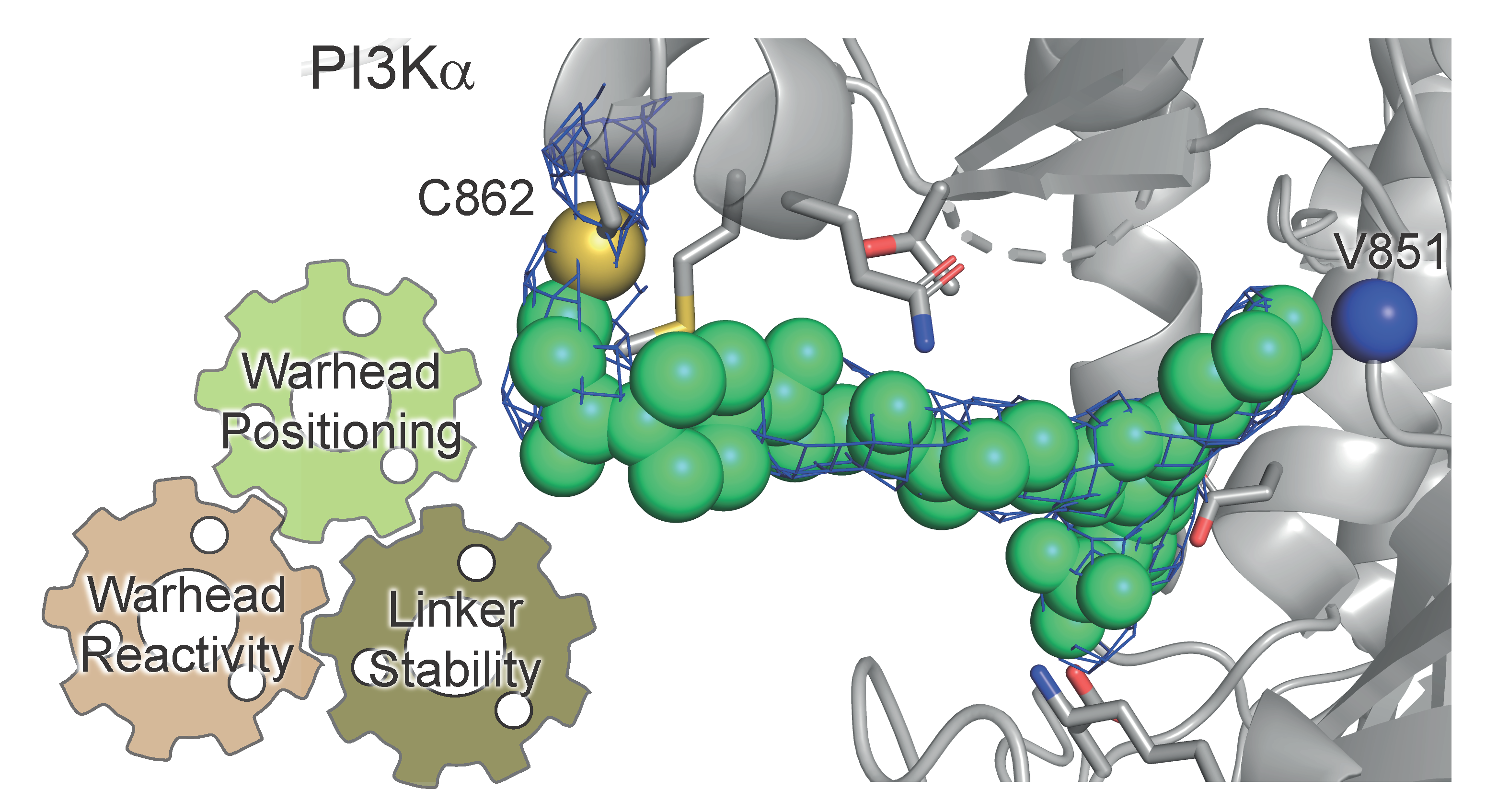
Herein we present a strategy to convert a phase II clinical candidate, a pan-PI3K inhibitor (PQR309, bimiralisib)[1,5], into highly selective PI3Kα-covalent inhibitors aiming to minimize the on-target metabolic side effects of PI3K inhibitor cancer therapy. We exploited a rational approach to increase target selectivity by covalently targeting a PI3Kα non-conserved nucleophilic amino acid side chain, namely Cys862. A reactive moiety, so called warhead, was introduced into a chemically modified bimiralisib.
A combination of warhead activity design, proximity and orientation allows a tight control of reversible inhibitor binding and isoform selective covalent binding. To avoid off-target reactions, we have set up a method to quantitatively evaluate warheads’ reactivity and optimize for selective Cys862 modification. An extensive Structure Activity Relationship (SAR) study was performed and a wide range of linear and restricted rotation linkers introduced. A comprehensive understanding of the kinetics of irreversible inhibition allowed to interpret SAR and identify compounds with optimal kinact (maximum potential rate of inactivation). X-ray crystallography and mass spectrometry experiments validated the covalent modification of Cys862. Our pilot compounds exceed specificity and potency over an experimental dimethyl-substituted enone, CNX-1351[6]. Moreover, they are metabolically stable in rat liver microsomes and outperform the rapidly metabolized CNX-1351.
Our strategy to investigate and tune warheads’ reactivity represents a major step forward in the rational design of covalent chemical tools. Moreover, we provide highly selective chemical tools to dissect PI3K isoform signaling in physiology and disease.
[1] Beaufils Florent et. al. J Med Chem. 2017, 60 (17), 7524-7538.
[2] Rageot Denise et. al. J Med Chem. 2019, 62 (13), 6241-6261.
[3] Borsari Chiara et. al. ACS Med Chem Lett. 2019, 10 (10), 1473-1479.
[4] Borsari Chiara et. al. J Med Chem. 2020, 63 (22), 13595-13617.
[5] Wicki Andreas et. al. Eur J Cancer. 2018, 96, 6-16.
[6] Nacht Mariana et. al. J Med Chem. 2013, 56 (3), 712-721.