How important are the residual nonadiabatic couplings for an accurate simulation of nonadiabatic quantum dynamics in a quasidiabatic representation?
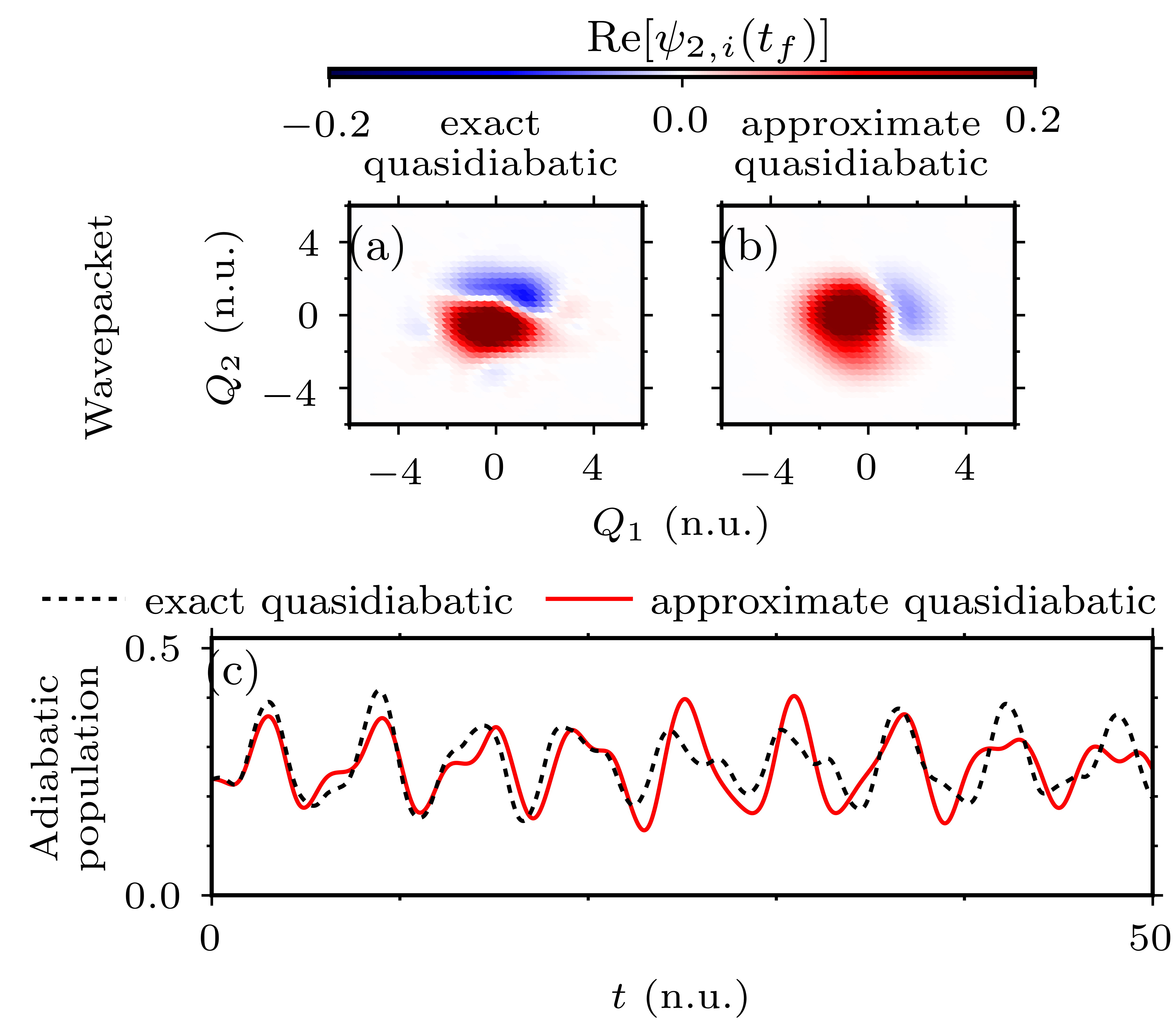
Diabatization of the molecular Hamiltonian is a standard approach to remove the singularities of nonadiabatic couplings at conical intersections of adiabatic potential energy surfaces. In general, it is impossible to eliminate the nonadiabatic couplings entirely—the resulting “quasidiabatic” states are still coupled by smaller but nonvanishing residual nonadiabatic couplings, which are typically neglected. Here, we propose a general method for assessing the validity of this potentially drastic approximation by comparing quantum dynamics simulated either with or without the residual couplings [1,2]. To make the numerical errors negligible to the errors due to neglecting the residual couplings, we use the highly accurate and general eighth-order composition of the implicit midpoint method [3,4]. The usefulness of the proposed method is demonstrated on nonadiabatic simulations in the cubic Jahn–Teller model of nitrogen trioxide and in the induced Renner–Teller model of hydrogen cyanide. We find that, depending on the system, initial state, and employed quasidiabatization scheme, neglecting the residual couplings can result in wrong dynamics. In contrast, simulations with the exact quasidiabatic Hamiltonian, which contains the residual couplings, always yield accurate results.
[1] S. Choi and J. Vaníček, J. Chem. Phys., 2021, 154, 124119.
[2] S. Choi and J. Vaníček, J. Chem. Phys., 2020, 153, 211101.
[3] S. Choi and J. Vaníček, J. Chem. Phys., 2019, 150, 204112.
[4] J. Roulet, S. Choi, and J. Vaníček, J. Chem. Phys., 2019, 150, 204113.